
Comparaison de chaînes ADN par programmation dynamique
Introduction
Il est possible de réaliser un alignement temporel entre deux chaînes ADN. Une des méthodes classiquement utilisées en informatique est la programmation dynamique présentée dès 1957 par Bellman ( Bellman 1957[1]) puis remaniée en 1978 par Sakoe et Chiba ( Sakoe et Chiba 1978[2]).
Méthode de Programmation Dynamique
Méthode
Soient
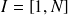 et
et
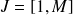 les supports temporels de
les supports temporels de
 et
et
Soit
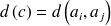 pour tout
pour tout
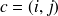 de
de
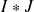 (distance locale)
(distance locale)Évaluer la dissemblance entre
et
revient à déterminer dans
un chemin
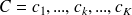 tel que
tel que
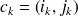
Les dissemblances entre A et B sont cumulées le long de ce fichier selon :
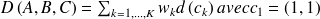 et
et
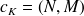 et
et
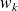 le poids attaché à l'arc
le poids attaché à l'arc
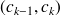
Les chemins C doivent vérifier les conditions suivantes :
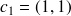 et
(coïncidence des extrémités)
et
(coïncidence des extrémités)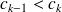 (monotonie : croissance temporelle)
(monotonie : croissance temporelle)continuité (i.e. pas de saut ou de « trou » sur le chemin)
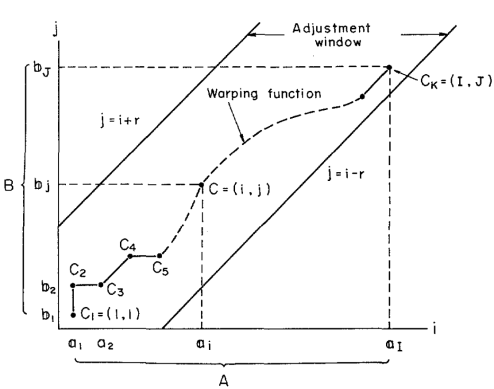
Contraintes locales et globales
Dans le domaine discret, il n'est pas possible d'introduire des contraintes de continuité : on introduit des notions de contraintes locales et globales qui permettent de limiter le nombre de chemins possible « autour » de la diagonale.
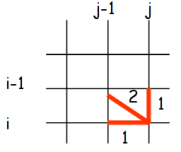
Exemple : Contraintes locales
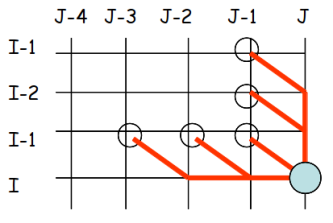
Pour accéder au point
 , on devra venir obligatoirement d'un des 5 points suivants :
, on devra venir obligatoirement d'un des 5 points suivants :
Exemple : Contraintes globales
On limite le chemin à une certaine enveloppe autour de la diagonale.
Il existe des pondérations symétriques :
le poids associé à un arc est la somme des progressions des deux indices
on convient que
 (poids de l'arc allant de
(poids de l'arc allant de
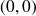 à
à
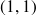 )
)
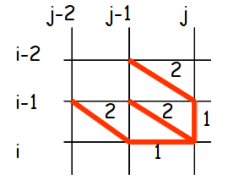
Exemple : Exemples de contraintes locales
Les
 sont les cumuls des
sont les cumuls des
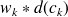 .
.
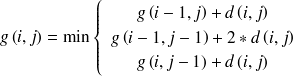
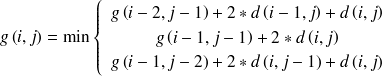
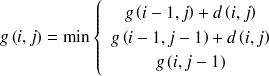
Comparaison optimale
La comparaison optimale est celle qui s'effectue le long d'un chemin
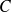 autorisé tel que
autorisé tel que
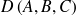 soit minimale, on note :
soit minimale, on note :
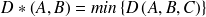 tel que
vérifie les conditions précitées.
tel que
vérifie les conditions précitées.
Le calcul se fait itérativement :
Tout chemin autorisé, allant de l'origine au point
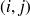 peut se décomposer en un 1er tronçon
peut se décomposer en un 1er tronçon
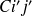 allant de l'origine au point
allant de l'origine au point
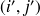 suivi d'un tronçon joignant
à
suivi d'un tronçon joignant
à
On peut donc écrire :
 cumul des
effectué entre les points
cumul des
effectué entre les points
 et
et
En ne considérant que les chemins passant par
on peut écrire :
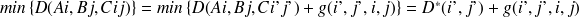
Cette formule permet de calculer par récurrence toutes les valeurs de
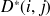 sur
sur
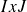 jusqu'à
jusqu'à
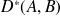
Complément : Un algorithme possible pour la programmation dynamique
Début
g(0,0) <-0
Pour (j=1,J) faire
g(O,j) <- +infini
Finpour
Pour (i=1,I) faire
g(i,0) <- +infini
Pour (j=1,J) faire
g(i,j) <- min{g(i-1,j)+d(i,j);
g(i-1,j-1)+2*d(i,j); g(i,j-1)+d(i,j)}
Finpour
Finpour
D=g(I,J)/(I+J)
Fin
Mise en œuvre
Vous allez mettre en œuvre cet algorithme. N'utilisez pas directement les bibliothèques disponibles en bio-informatique, on vous demande ici de programmer cet algorithme sur des séquences ADN. Les demandes formulées ci-dessous sont en ordre croissant de difficulté.
Vous pouvez tester vos algorithmes sur les séquences que vous avez jusqu'à présent utilisé, ou bien en utiliser d'autres, de taille différente afin de pouvoir réaliser l'alignement. Si les deux séquences ADN sont trop différentes, les résultats ne seront pas très pertinents.
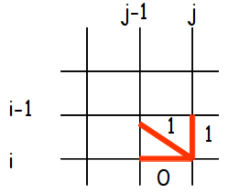
Contraintes locales simples
Vous allez mettre en place l'algorithme en considérant que la distance entre deux base identique est égale à 0 et 1 sinon. La contrainte locale à utiliser est décrite dans la figure ci dessous.
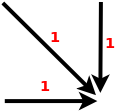
Calculez la matrice de coût et déterminez le coût grlobal (normalisé par la longueur des séquences).
Affichez ces informations (ou une partie de ces informations si les séquences sont trop longues : juste le début ou la fin de la matrice) en format texte à l'écran.
Affichage du meilleur chemin
Calculer le meilleur chemin entre les deux séquences. Pour cela vous pouvez reparcourir votre matrice de coûts à l'envers en recherchant le minimum des valeurs adjacentes. Affichez le les coordonnées des points du meilleur chemin. Vous pouvez également afficher les séquences et symboliser les insertions et suppressions sous forme de caractères d'espacement.
Veuillez également fournir des statistiques sur :
le nombre de bases attendues : T
le nombre de bases en corcondance : C
le nombre de substitutions : S
le nombre d'insertions : I
le nombre de suppressions : D
le taux d'exactitude :
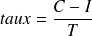
Exemple : Exemple d'alignement temporel entre deux séquences
ref : ACCGGGC___TACGA
hyp : ACC___CTTTTAGCA
dans cet alignement : GGG a été supprimé et TTT a été inséré. C a été remplacé par G et G par C à la fin.
utilisation de contraintes globales
Reprenez votre algorithme et rajoutez la prise en compte de contraintes globales : c'est à dire l'impossibilité de passer par les extrémités de la matrice de coût. Interdisez les passages par les cases qui s'éloignent à plus de 10 cases de la diagonale ("adjustement window" dans la figure présenant la méthode de programmation dynamique).
Utilisation de contraintes locales plus compliquées et affichage de l'alignement
Reprenez votre algorithme en utilisant des contraintes locales plus complexes :
en ne prenant pas tous les poids à 1 (attention, celà aura une conséquence sur la recherche du meilleur chemin, vous serez amené à revoir cette partie)
en utilisant des contraintes locales plus distantes
en utilisant des contraintes locales asymétriques.
Distances entres bases
Est-ce que la distance entre une base et une autre doit être uniforme ? Cherchez dans la littérature les distances et justifications qui peuvent être choisies pour justifier des distances autre. Reprenez vos programmes avec ces hypothèses.




